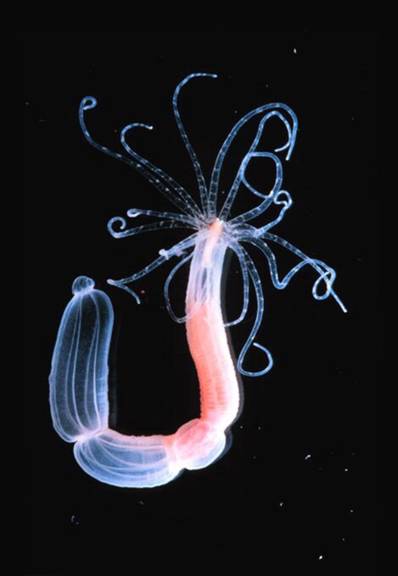
Nematostella vectensis

Bicyclus anynana

Drosophila melanogaster
The project is financed by the EU Complexity Net programme (Netherlands Organisation for Scientific Research, NWO, the Portugese Science Foundation (FCT) and the Spanish Science Foundation (MICINN)
One of the most spectacular developments in modern biology is the discovery of gene regulatory networks involved in pattern formation during embryo development (Davidson 2006). An important next step is to understand the regulatory structure and dynamics of these networks; this will lead to fundamental new insights in developmental biology, evolution and eventually to new medical applications. Disruptions in regulatory networks play an important role in developmental malformations and diseases such as cancer. Most of these networks are characterized by very complex regulatory structures. An additional difficulty is that network dynamics vary across space and time and are influenced by biomechanical events (e.g.formation of cell layers, cell migration, cell death, and cell division). A simple graph representation showing the connections between genes does not provide insight into the dynamical behaviour of such complex regulatory systems. Instead, detailed dynamical models will be required. However, obtaining such models is not straightforward. First of all, it is often not clear which network-modelling formalism should be used. Due to the biochemical complexity of eukaryotic transcription, it is currently impossible to derive network models from first principles (Reinitz et al., 2003). Instead, many phenomenological formalisms have been used, from Boolean models to differential equation models approximating transcriptional dynamics with sigmoid functions to stochastic formalisms (de Jong, 2002). There are currently no widely applicable guidelines for the choice of model, which depends on the specific problem under study, as well as the availability and quality of expression data. Another important challenge is the determination of model parameters. Even models of moderately sized networks contain a large number of parameters, which determine production, diffusion and decay rates as well as regulatory interactions or the regulatory topology of a gene network. These parameters are often difficult (if not impossible) to measure. Instead, they have to be inferred by fitting models to data using global, non-linear optimization (Fig. 1). This type of reverse-engineering approach poses a number of significant challenges. One major issue for parameter inference is that the observed dynamical behaviour of the system can often be explained by distinct regulatory mechanisms. This can be due to the optimization problem being ill-posed or insufficiently constrained by data (Ashyraliyev et al., 2009a). Alternatively, parameters can be difficult to determine due to correlations between them (Gutenkunst et al., 2007; Ashyraliyev et al., 2008; 2009b). Model validation based on additional experimental evidence is required to decide, which of the alternative mechanisms is applicable to the real biological system. This is often timeconsuming and technically challenging. Therefore, it is essential to decrease the number of alternative predictions that need to be tested experimentally. One way of achieving this is to take additional criteria into account for model fitting. Usually, the accuracy with which a model reproduces observed expression patterns is measured by a cost function based on the sum of squared differences between model and data (single-objective optimisation). Here, we propose to take advantage of the fact that pattern formation must proceed reliably in the presence of molecular fluctuations, genetic variability and environmental perturbations. In other words, realistic patterning mechanisms are robust, and robustness should be considered when fitting models to data. This is achieved by adding additional optimisation criteria to the fitting procedure (multi-objective optimisation; see Fig. 1 and Handl et al., 2007). Preliminary efforts have been made to apply multi-objective optimisation to reverse-engineering gene networks (van Someren et al., 2003; Esmaeili et al., 2009; Guo et al., 2009), but none of these efforts have yet been applied to real developmental systems.
In this project, we propose to use multi-objective optimisation for a careful comparison of the performance of three distinct network modelling formalisms applied to the study of real-world developmental gene regulatory networks. We will compare a connectionist (gene circuit) formalism with models based on Hill-functions, or the law of mass action. Models will be optimised both with regard to their ability to fit the data, as well as their robustness towards molecular fluctuations and changes in parameter values. For model fitting we will use qualitative and quantitative spatial gene expression data from three species (the sea anemone Nematostella vectensis, the butterfly Bicyclus anynana, and the fruit fly Drosophila melanogaster), which vary vastly in their quality, coverage and resolution. This allows us to test systematically under which circumstances each formalism is best able to reproduce the observed expression patterns, is most suited for prediction of network topology and mutant (or otherwise perturbed) gene expression, and is able to reproduce the robustness of pattern forming gene regulatory networks.
The central research question we propose to investigate in this project is to characterize the genes that control the differences in coral morphology for related coral species. We do this by a quantitative comparison of gene expression patterns. We especially focus on genes involved in the process of calcification. To test the hypothesis that these genes can explain the differences in morphology, we plan to use the estimated quantities in a simulated network controlling calcification. We want to study the emergence of the micro-morphology structure and link gene expression patterns to the corallite structure. This polyp (corallite) based model will be coupled with a macroscopic growth form model describing Ca2+ and HCO3- fluxes from the environment. A better understanding of calcification in corals is of fundamental importance in research on the potentially detrimental impact of increasing atmospheric carbon dioxide concentrations, reducing ocean pH and carbonate ion concentrations on the calcification process in corals and other calcifying organisms.